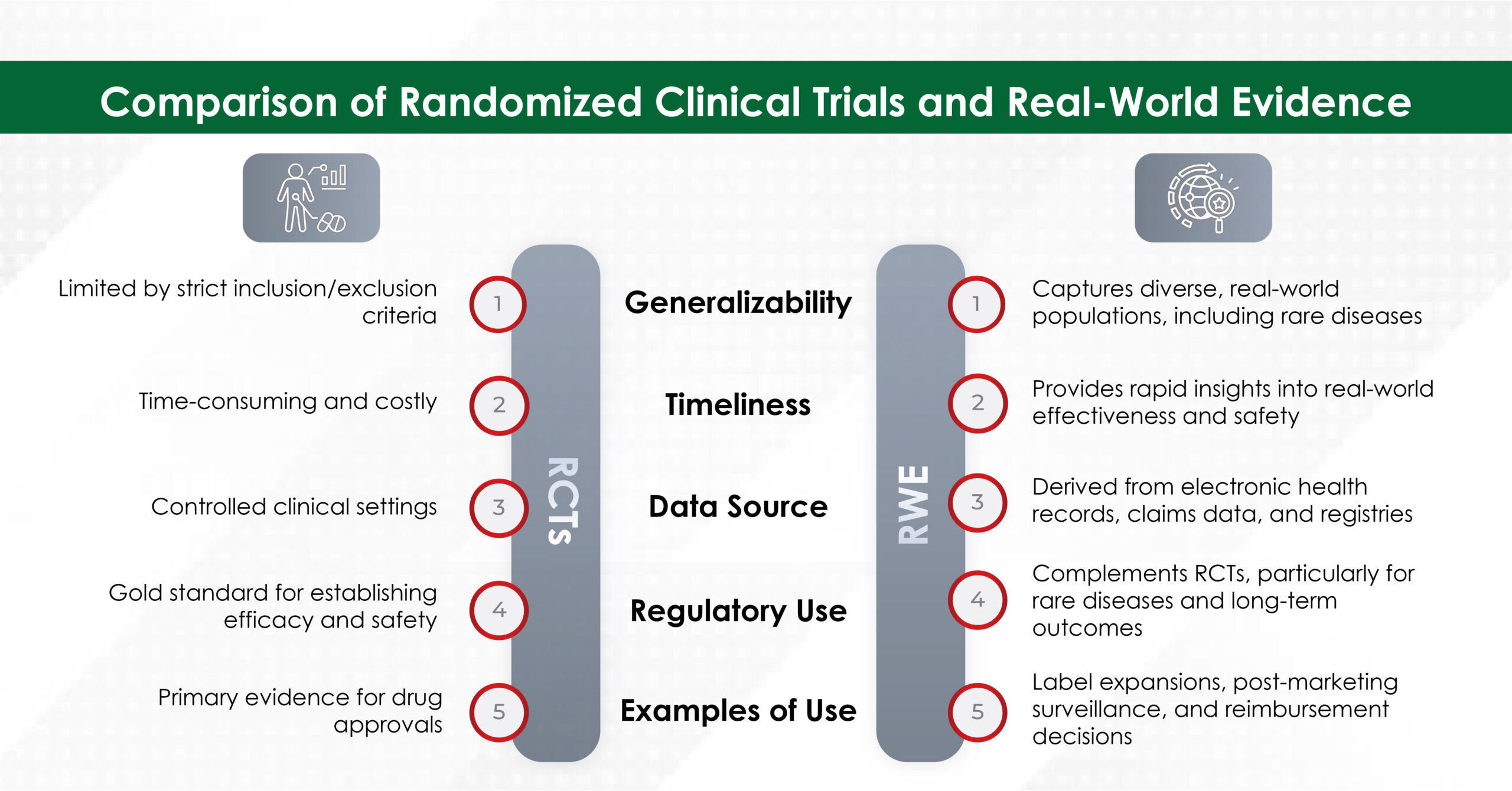
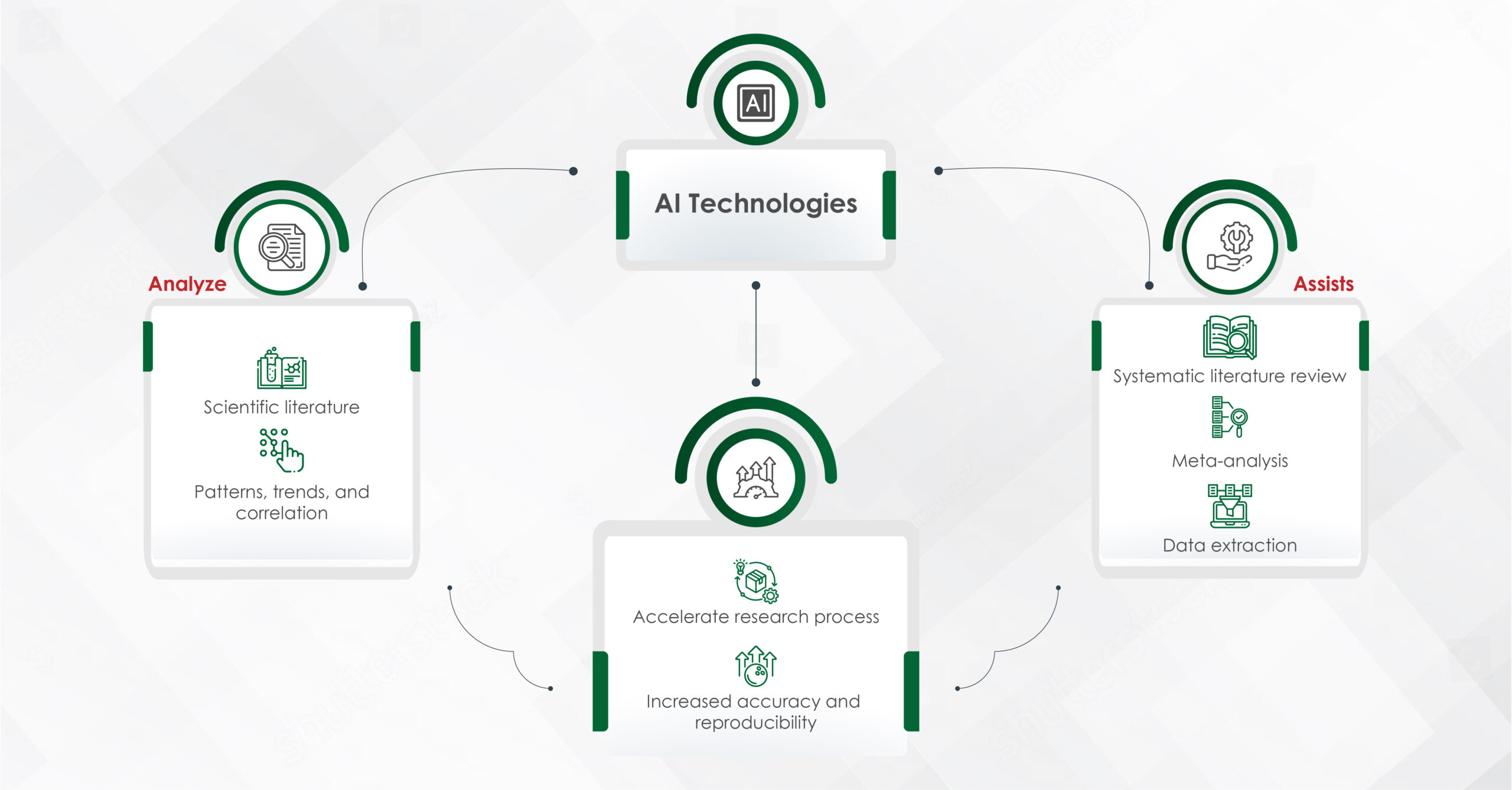
In an era where speed, efficiency, and personalization have become imperatives, Artificial Intelligence (AI) — particularly machine learning, AI algorithms, and generative AI — is revolutionizing how the pharmaceutical industry approaches content creation. From automating reference linking to generating first drafts, AI is making content smarter, faster, and more scalable.
Yet, amidst this digital acceleration, one thing remains clear that human expertise is still the foundation of trust in healthcare communication.
At recent industry events and conferences, thought leaders echoed a common sentiment: AI is a catalyst, not a replacement. The most impactful pharma content today is born from a powerful synergy between AI and human intelligence, particularly when applied across disciplines such as data analysis, pharmacovigilance, clinical trials, and drug development.
The power of AI in pharma content creation
AI has significantly shortened the timeline of content development. By automating routine tasks like data architecture, extraction, summarization, and even modular content creation, AI enables teams to focus on higher-value activities. Some of the key advantages include:
- Speed and efficiency: Drafting scientific summaries, creating modular content blocks, and auto-tagging assets for reuse — AI can perform these tasks in minutes, which previously took days.
- Compliance and consistency: AI can cross-reference regulatory guidelines and validate claims faster, ensuring fewer errors and a higher level of consistency across materials.
- Personalization at scale: Machine learning and AI algorithms help in tailoring content to specific audiences, ensuring that messaging is relevant, targeted, and timely – particularly in fields like personalized medicine and patient care.
The impact is undeniable. Pharma companies are no longer just creating more content — they are creating better content, delivered at the right time, to the right audience.
Why humans remain at the heart of trusted communications
Yet, even as AI transforms the operational side of content creation, human oversight remains indispensable for several reasons:
- Contextual accuracy and nuance
Medical communication is not just about relaying information — it’s about conveying it with precision, empathy, and context. AI can process data, but only humans can interpret complex medical nuances, drug discovery insights, and cultural considerations that shape the right narrative.
- Ethical and regulatory oversight
In the field of regulatory pharmalike pharmacovigilance and clinical trials, accuracy and ethics are non-negotiable. While AI can check references, it cannot assure about the ethical gray areas – a responsibility that remains with human experts.
- Building emotional connection
Trust in healthcare communications is deeply emotional. Physicians, patients, and stakeholders seek authenticity and human connection – something machines like large language models cannot replicate. Skilled writers infuse compassion, clarity, and credibility into every piece of content, building the trust that AI alone cannot establish.
- Innovation and strategic thinking
AI can optimize existing processes but cannot create disruptive strategies. Human creativity is still essential in commercial areas like customer engagement, content marketing, and long-term drug development strategies.
The future is Human + Machine, Not Human vs Machine
The future of pharma content creation lies in collaboration, not competition. AI — whether used in pharma AI tools, generative AI, or data analysis pipelines — should be seen as a powerful tool that augments human potential. When medical writers, regulatory experts, and creative strategists partner with AI, the result is content that is faster, smarter, and — most importantly — trusted.
At Turacoz, we believe that technology is only as powerful as the people who wield it. By combining the scalability of AI with the critical thinking, empathy, and expertise of humans, we help our clients craft communications that build lasting trust with healthcare professionals and patients alike.
Final thoughts
AI is redefining the way content is created in pharma, making processes smarter, faster, and more scalable — especially with advancements in AI algorithms, data architecture, and personalized medicine. But even the most sophisticated AI cannot replace the human touch that drives trust, credibility, and connection in healthcare communications.
As we embrace this exciting new era, the winning formula is clear: leverage the best of AI innovation, anchored by the irreplaceable value of human expertise.
Ready to elevate your content strategy with the perfect blend of human insight and AI innovation? Connect with Turacoz today.